Cystic fibrosis (CF) is an inherited, life-threatening disorder that damages the lungs and digestive tract. It is caused by a defective gene that triggers the production of thickened mucus that clogs airways and blocks the secretion of digestive enzymes. People with CF have a mutation in the CFTR gene, which leads to altered function of a protein that controls the movement of salt and water in and out of cells. CF causes thick, sticky mucus to build up in the lungs, which can lead to serious respiratory problems. The CFTR protein is a channel protein that controls the flow of H2O and Cl− ions in and out of cells inside the lungs. When the CFTR protein is working correctly, ions freely flow in and out of the cells. However, when the CFTR protein is malfunctioning, these ions cannot flow out of the cell due to a blocked channel. This causes cystic fibrosis, characterized by the buildup of thick mucus in the lungs.
Symptoms are progressive and often severe, and they may include breathing problems, recurrent lung infections, poor growth, male infertility, and chronic inflammation of the pancreas, liver, kidneys, and heart.
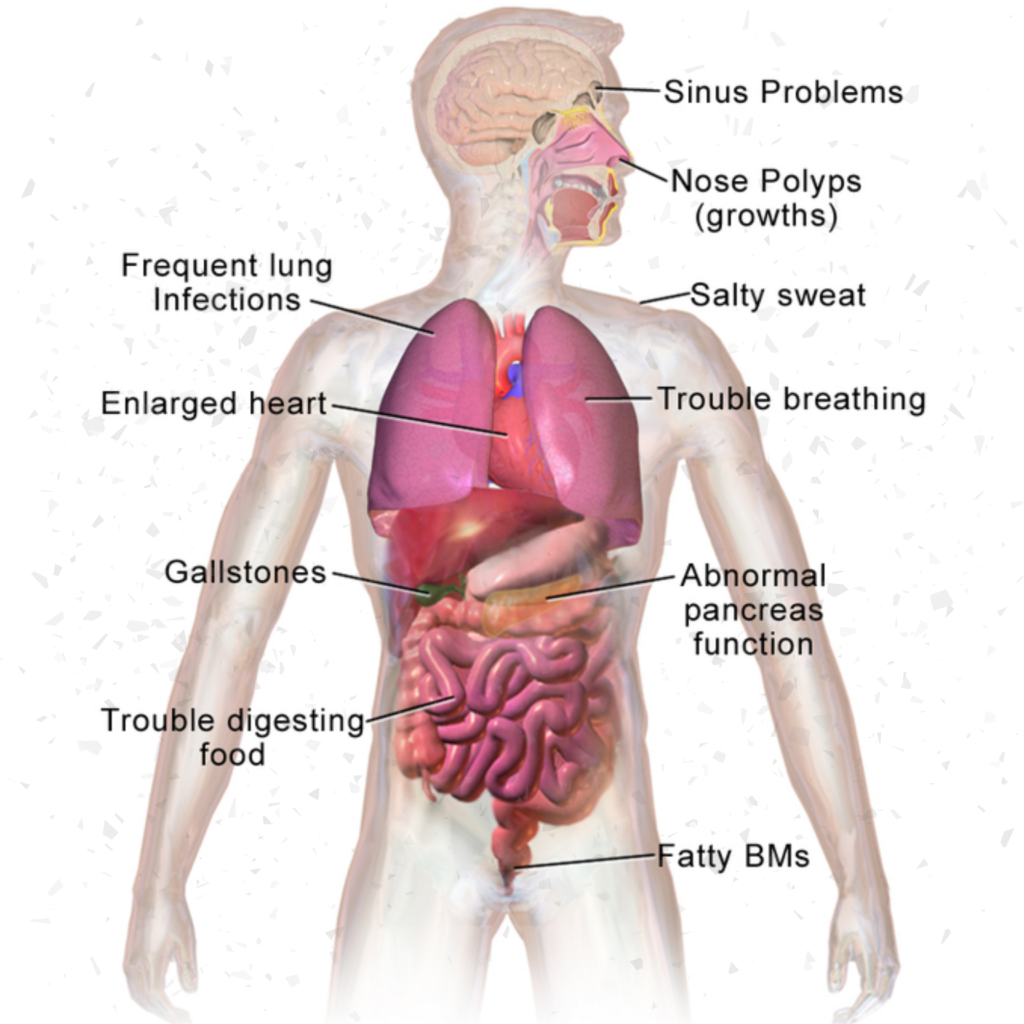
The mucus accumulates in the organs and clogs them, preventing them from doing their jobs correctly. In the lungs, the build up of mucus can lead to respiratory problems such as irreversible lung damage due to infection, airway blockage which leads to difficulty breathing, chest infections and coughing for large portions of life.
The mucus also can block the pancreatic ducts, preventing digestive enzymes from reaching the intestine and leading to malnutrition. The mutations lead to thick mucus being secreted from the glands throughout the body, especially within the lungs, pancreas, liver, intestines and reproductive system. Treatment options include antibiotics to treat lung infections, airway clearance techniques to help remove mucus from the lungs, and nutrition therapy to ensure adequate intake of calories and nutrients. In some cases, surgery may be needed to clear blocked pancreatic ducts. With appropriate treatment, many people with CF can lead relatively normal lives. If the CFTR gene mutation occurs in the pancreas, however, it can lead to malnutrition and digestive problems such as diarrhoea and steatorrhoea (fatty, offensive-smelling stool).
There is no cure for CF, but treatments are available to help manage the symptoms. Treatment options include antibiotics to treat lung infections, airway clearance techniques to help remove mucus from the lungs, and nutrition therapy to ensure adequate intake of calories and nutrients. In some cases, surgery may be needed to clear blocked pancreatic ducts.
With appropriate treatment, many people with CF can lead relatively normal lives. In fact, the median predicted age of survival for people with CF is close to 40 years. However, many people with CF die young. Cystic fibrosis typically manifests early in life. Newborns and infants with cystic fibrosis tend to have frequent, large, greasy stools (a result of malabsorption) and are underweight for their age. 15–20% of newborns have their small intestine blocked by meconium, often requiring surgery to correct.[11] Newborns occasionally have neonatal jaundice due to blockage of the bile ducts. Children with cystic fibrosis lose excessive salt in their sweat, and parents often notice salt crystallizing on the skin, or a salty taste when they kiss their child. The primary cause of morbidity and death in people with cystic fibrosis is progressive lung disease, which eventually leads to respiratory failure.
Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation. In later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing. Other signs include high blood pressure in the lung (pulmonary hypertension), heart failure, difficulties getting enough oxygen to the body (hypoxia), and respiratory failure requiring support with breathing masks.
In addition to typical bacterial infections, people with CF more commonly develop other types of lung diseases. Among these is allergic bronchopulmonary aspergillosis, in which the body’s response to the common fungus Aspergillus fumigatus causes worsening of breathing problems. Another is infection with Mycobacterium avium complex, a group of bacteria related to tuberculosis, which can cause lung damage and do not respond to common antibiotics.
Mucus in the paranasal sinuses is equally thick and may also cause blockage of the sinus passages, leading to infection.
This may cause facial pain, fever, nasal drainage, and headaches. Individuals with CF may develop overgrowth of the nasal tissue (nasal polyps) due to inflammation from chronic sinus infections. Recurrent sinonasal polyps can occur in 10% to 25% of CF patients.[16]: 1254 These polyps can block the nasal passages and increase breathing difficulties.
CF can be diagnosed with blood tests, genetic screening, and a procedure known as a sweat chloride test. While there is no cure for CF, there are treatments that can improve both the length and quality of one’s life.